Теоретическая модификация γ-мангостана через окисление пренильной группы до карбоксильной для повышения эффективности сенсибилизатора в цвето-сенсибилизированных солнечных элементах (DSSC)
Теоретическая модификация γ-мангостана через окисление пренильной группы до карбоксильной для повышения эффективности сенсибилизатора в цвето-сенсибилизированных солнечных элементах (DSSC)

Аннотация
Природные красители стали недорогой и экологически безопасной альтернативой синтетическим сенсибилизаторам в цвето-сенсибилизированных солнечных элементах (DSSC), хотя их эффективность по-прежнему остается относительно низкой. γ-Мангостан является одним из основных ксантоновых пигментов в кожуре мангостина и демонстрирует многообещающие донорные свойства, однако его эффективность ограничивается отсутствием фиксирующей группы для прочной адсорбции на поверхности полупроводника. Целью данного исследования является улучшение его фотоэлектрических свойств через теоретическую модификацию путем окисления пренильной боковой цепи в карбоксильную (–COOH) группу с использованием вычислительных методов DFT и TD-DFT (B3LYP/6-311+G**). Результаты показывают, что модификация снижает энергетические уровни HOMO и LUMO и усиливает внутримолекулярный перенос заряда, что приводит к расширению пиков поглощения света в сторону более длинных волн. После замещения карбоксильной группой параметр электронной связи |VRP| увеличивается с 0,82 до 0,96 эВ, в то время как эффективность улавливания света (LHE) повышается с 0,32 до 0,53, что указывает на усиление способности к введению электронов и поглощению фотонов. Хотя величина ΔGinject незначительно увеличивается (-2,85 эВ по сравнению с -3,30 эВ), общие результаты показывают, что γ-мангостан+COOH является более эффективным кандидатом в красители, чем природный γ-мангостан. Данная работа предоставляет теоретические доказательства для разработки структурно модифицированных натуральных сенсибилизаторов, обеспечивающих более высокую эффективность DSSC.
1. Introduction
Dye-sensitized solar cells (DSSCs) offer a promising photovoltaic technology owing to their simple fabrication, relatively low production costs, and adaptability to various light conditions. Natural dyes have gained increasing attention as sustainable alternatives to synthetic sensitizers due to their low toxicity and abundance . Despite these advantages, natural dyes often suffer from poor chemical stability, limited anchoring capability, and narrow absorption ranges, resulting in lower efficiencies compared with synthetic metal complexes and organic dyes .
Mangosteen (Garcinia mangostana) pericarp extract is among the most widely studied natural dye sources for DSSCs. It contains xanthone derivatives—α-, β-, and γ-mangostin—that exhibit conjugated aromatic structures capable of absorbing UV–visible radiation . Among these, γ-mangostin shows the greatest electron-donating potential and has been reported to contribute significantly to observed photovoltaic activity. Extract-based DSSCs have achieved efficiencies ranging from 1,17% to 1,78% through solvent optimization . However, these values remain much lower than the >10% efficiencies routinely obtained using synthetic dyes.
A key structural drawback of γ-mangostin is the absence of an anchoring group, such as –COOH, –H₂PO₃, or –SO₃H, which is essential for strong adsorption onto TiO₂ surfaces and efficient electron injection . The prenyl group on γ-mangostin is susceptible to oxidation, allowing conversion to carbonyl or carboxyl functionalities . Introducing a carboxyl group is expected to improve binding strength, enhance electronic communication with TiO₂, and potentially expand the absorption spectrum . Previous theoretical studies have shown that electron-withdrawing substituents, such as rhodanine-acetic acid, significantly improve γ-mangostin’s DSSC-related parameters .
Computational chemistry, particularly DFT and TD-DFT, has become indispensable for predicting electronic structures, charge-transfer behavior, and photovoltaic descriptors relevant to dye performance. Novir et al. (2017) demonstrated that B3LYP/6-311+G** provides reliable HOMO–LUMO energies, ΔGinject, electronic coupling constants (|VRP|), and light-harvesting efficiencies (LHE) for organic dyes .
Despite the potential of structural modification, no study to date has examined γ-mangostin prenyl-to-carboxyl oxidation from a theoretical perspective. Considering that Indonesia is a major mangosteen producer , the utilization of mangosteen pericarp—typically discarded as waste—as a renewable DSSC material is of national relevance and global interest.
This work aims to computationally assess whether introducing a –COOH anchoring group improves the photovoltaic characteristics of γ-mangostin. The insights gained are expected to support the rational design of high-performance natural sensitizers.
2. Methods
2.1. Theoretical Background
The primary metric for evaluating the photovoltaic performance of Dye-Sensitized Solar Cells (DSSCs) is the power conversion efficiency (η). It is determined by the following formula :
In this equation, VOC represents the open-circuit photovoltage, JSC denotes the short-circuit photocurrent density, FF is the fill factor, and Pinc is the intensity of incident sunlight.
To improve η, it is necessary to enhance JSC, which can be estimated by integrating the following parameters :
where LHE(λ) represents the light harvesting efficiency at maximum wavelength, ϕinject is the efficiency of electron injection, and ηcoll is the efficiency of electron collection, which only relate to the architecture of DSSC. The light harvesting efficiency (LHE) is further defined as :
Here, f represents the oscillator strength of the dye associated with the absorption maximum λmax. It is found that the higher ƒ value will produce the higher LHE for the sensitizer and therefore the higher value of JSC.
The efficiency of electron injection ϕinject is a function of the driving force ΔGinject. A larger ΔGinject value typically results in a higher JSC. This driving force for the transition from the excited organic dye to the TiO2 semiconductor conduction band is calculated as :
In this equation, Edye is the excited state oxidation potential, and is the reduction potential of the TiO
where Edye is the ground state oxidation potential (estimated as the negative EHOMO of the dye) and Eex is the electronic vertical transition energy related with λmax.
The performance is also influenced by the dye regeneration driving force ΔGregn, calculated by :
where Eredox is the I⁻/I₃⁻ potential of the redox couple (4,8 eV). Additionally, the coupling constant VRP affects the electron injection rate. It is derived from the following equations :
From this equation, it is found that the higher ΔERP would lead to enhanced the |VRP| which would increase the electron injection in DSSCs. The ΔERP can be calculated by Koopmans approximation as :
For efficient charge transfer, the electron injection time must be shorter than the decay time to the ground state. Dyes with a longer excited-state lifetime (τ) minimize charge recombination. This parameter is calculated using Einstein transition probabilities :
where ΔE is the first excitation energy of the dye.
2.2. Molecular Design
Structures of γ-mangostin and γ-mangostin+COOH were constructed using Avogadro 1.2.0 and exported in ORCA (.inp) format (Fig. 1).
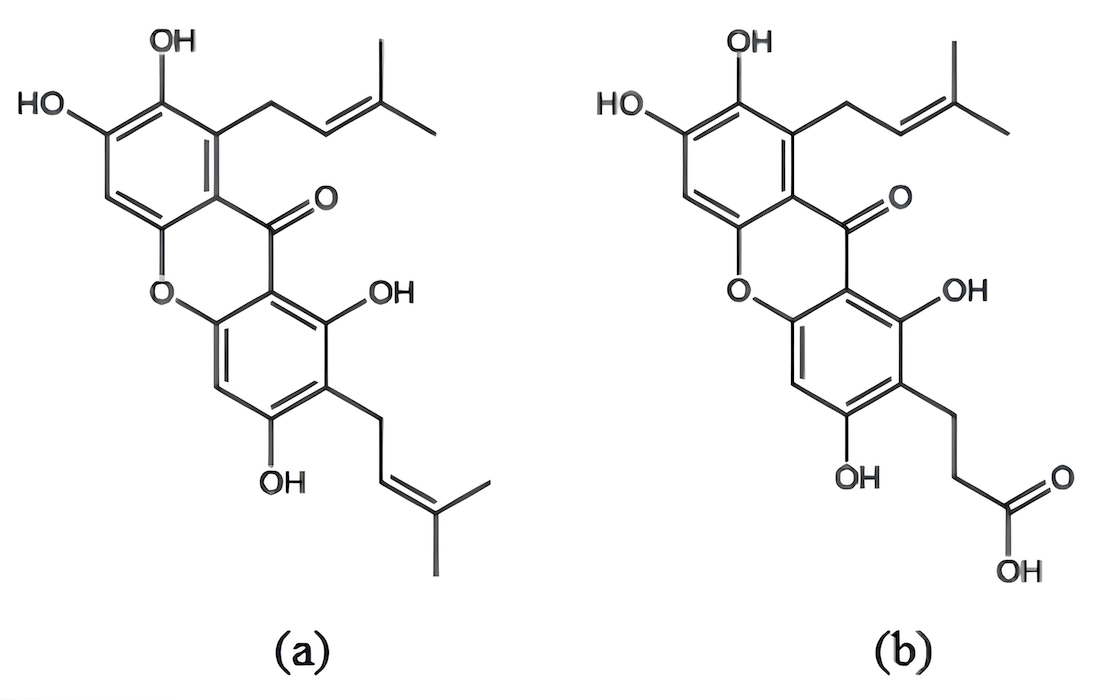
Molecular structures of γ-mangostin (a) and γ-mangostin+COOH (b)
It should be noted that explicit dye–TiO₂ interfacial modeling, such as adsorption geometry optimization, binding energy evaluation, or surface charge-transfer analysis, was not performed in this study. Therefore, dye–TiO₂ binding strength and anchoring configurations were not directly quantified. Any discussion related to surface anchoring is inferred from molecular electronic structure descriptors and established DSSC design principles, and should be regarded as indicative rather than conclusive.
2.3. Geometry Optimization and Electronic Properties
All geometries in this work were optimized use ORCA with DFT-B3LYP/6-311+G**, followed by TD-DFT calculations at the same level to evaluate electronic properties. All calculations were performed in the gas phase to isolate the intrinsic electronic effects of molecular modification. In practical DSSC environments, solvent polarity, electrolyte composition, dye protonation state, and surface adsorption can significantly shift absolute orbital energies and electron-injection thermodynamics. Therefore, absolute energy alignments and ΔGinject values reported here should be interpreted qualitatively. Nevertheless, gas-phase calculations remain suitable for evaluating relative trends between structurally related dyes, which is the primary focus of this study.
3. Results and Discussion
3.1. HOMO–LUMO Analysis
HOMO and LUMO energy levels are essential parameters for evaluating molecular stability and the thermodynamic feasibility of electron transfer. As shown in Figure 2, both γ-mangostin and γ-mangostin+COOH meet the energy alignment requirements for electron injection in DSSCs. Electron transfer proceeds thermodynamically from the dye’s LUMO to the conduction band (CB) of TiO₂, then to the external circuit, and finally back to the dye’s HOMO.
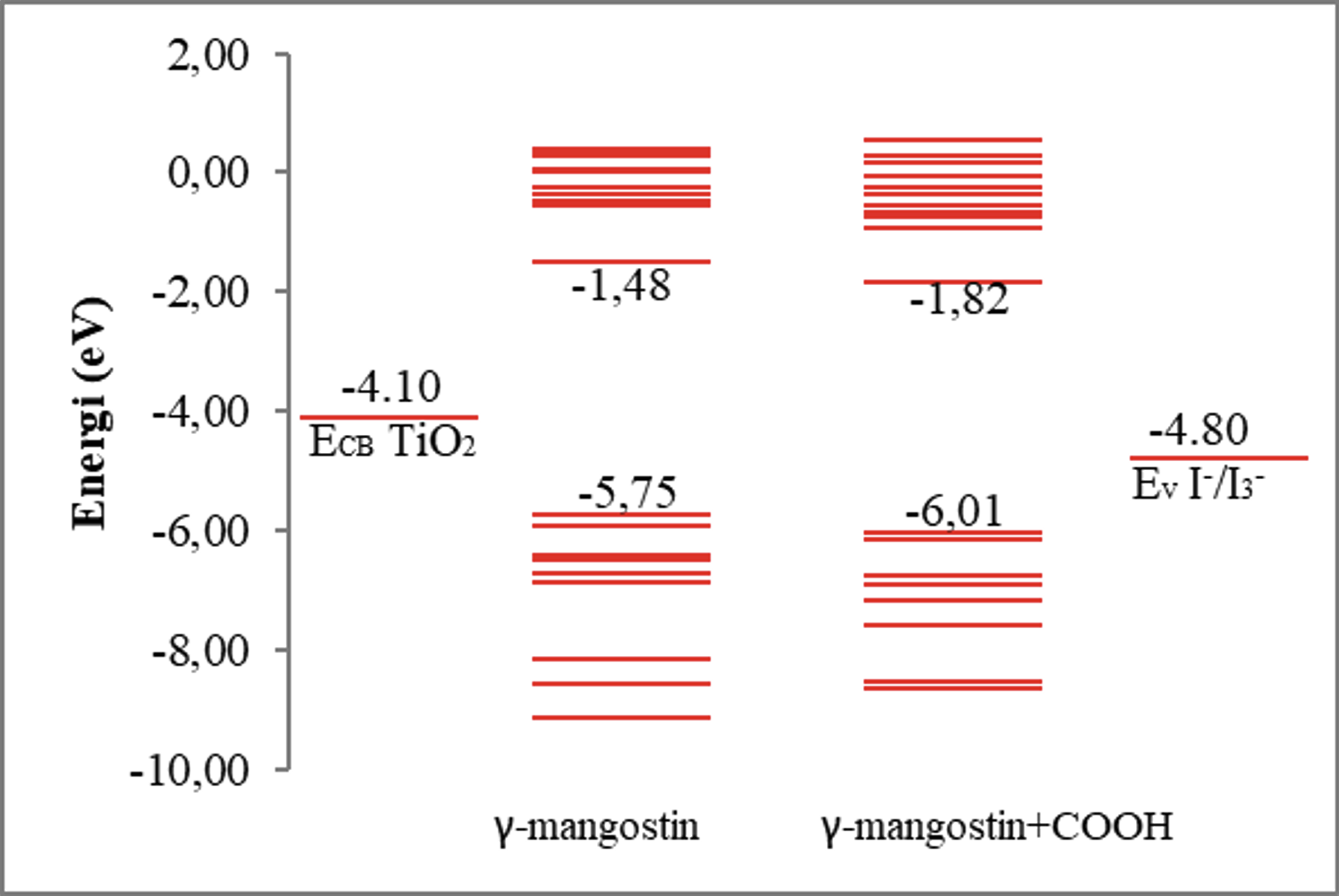
HOMO–LUMO energy levels of γ-mangostin and γ-mangostin+COOH
Electron regeneration from the electrolyte to the oxidized dye must also be thermodynamically favorable. This requires the dye’s HOMO energy to lie below the redox potential of the I⁻/I₃⁻ electrolyte (-4,80 eV) . γ-Mangostin (-5,75 eV) and γ-mangostin+COOH (-6,01 eV) both meet this requirement, providing spontaneous driving forces of -0,95 eV and -1,21 eV, respectively.
The introduction of a –COOH group not only provides anchoring capability but also acts as an electron-withdrawing substituent. This effect is reflected in the decrease of the HOMO energy from -5,75 to -6,01 eV, which enhances the driving force for dye regeneration. A more negative regeneration driving force accelerates the recovery of oxidized dye molecules, reducing the likelihood of charge recombination with electrons in TiO₂ .
3.2. UV–Vis Spectral Characteristics
The experimental UV–Vis spectrum of γ-mangostin as shown in Figure 3 displays three main absorption regions, characteristic of the xanthone chromophore. The band at 243 nm corresponds to a π→π* transition within the conjugated C=C framework. A shoulder at approximately 265 nm is associated with n→σ* transitions originating from ether (C–O–C) groups. At longer wavelengths, absorptions between 317–352 nm are attributed to n→π* transitions of carbonyl groups (C=O), consistent with typical xanthone electronic behavior. The broad extension to 352 nm indicates spectral widening due to interactions between oxygen lone-pair orbitals and the delocalized π-system, suggesting an extended conjugation network .
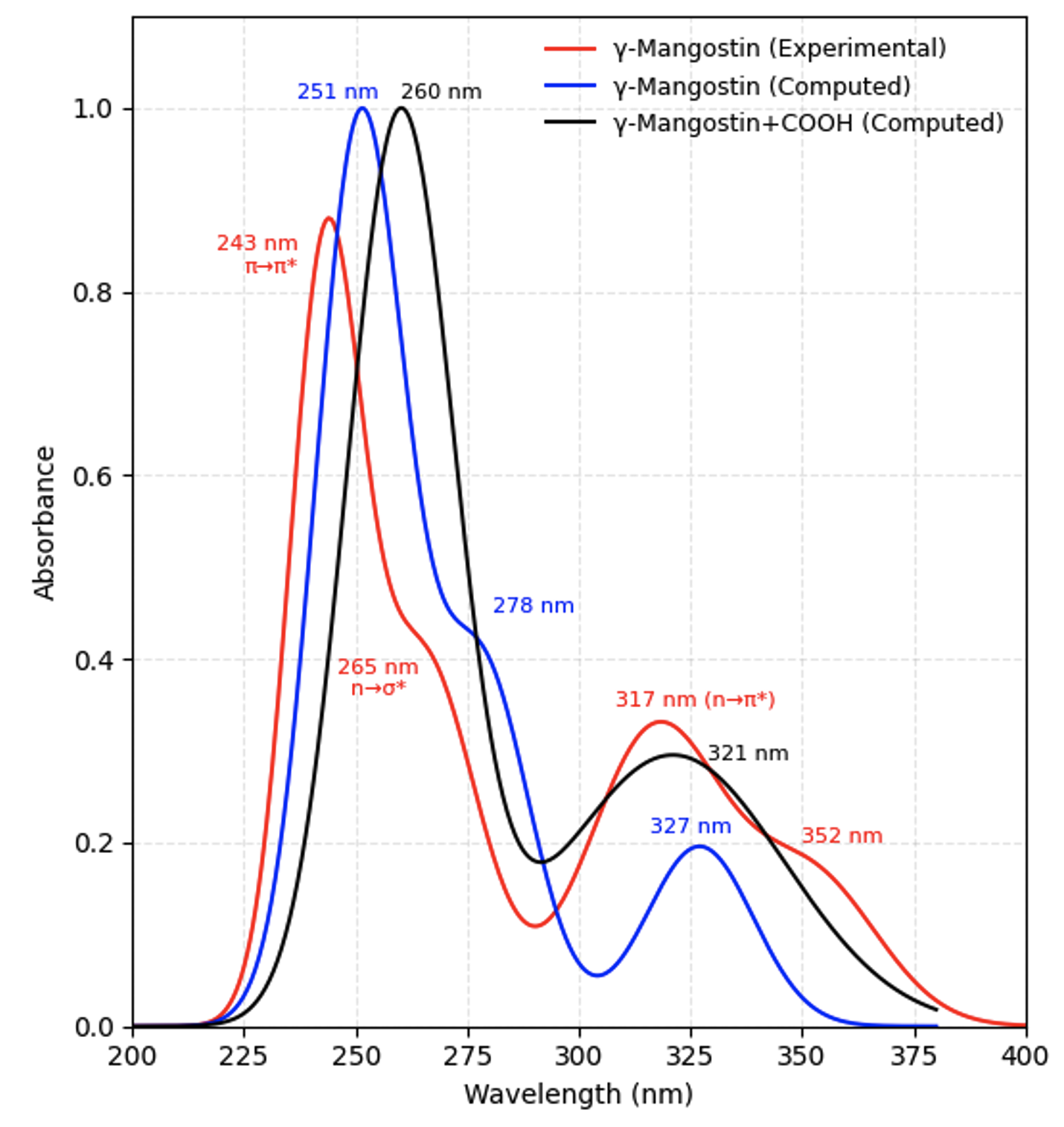
UV-Vis spectra of γ-mangostin (experimental), γ-mangostin (computed), and γ-mangostin+COOH (computed)
Figure 3 compares the experimental spectrum with TD-DFT (B3LYP/6-311+G**) computational results. The simulated spectrum of γ-mangostin (blue curve) aligns well with experimental features, exhibiting major bands at 251 nm and a secondary band around 278 nm. Excited-state analysis shows that the 251 nm band arises primarily from the HOMO-1 → LUMO+1 transition (53%), while the 278 nm band is governed by the HOMO-4 → LUMO transition (39%) as summarized in Table 1. A lower-energy excitation at 327 nm originates from a mixture of HOMO-2 → LUMO (37%) and HOMO → LUMO (85%) transitions, confirming the direct involvement of frontier orbitals.
Percentage contributions of electronic transitions for γ-mangostin and γ-mangostin+COOH
Dye | λ, nm | Transition Type | Contribution, % |
γ-mangostin | 251 | HOMO-1 → LUMO+1 | 53 |
278 | HOMO-4 → LUMO | 39 | |
327 | HOMO-2 → LUMO | 37 | |
HOMO → LUMO | 85 | ||
γ-mangostin+COOH | 260 | HOMO-1 → LUMO+1 | 34 |
321 | HOMO-1 → LUMO | 64 |
Upon the introduction of a –COOH group (black curve), the primary absorption band shifts to 260 nm, accompanied by the emergence of a new band near 321 nm. This modification alters both orbital energies and excitation characteristics. The 260 nm band is dominated by a HOMO-1 → LUMO+1 transition (34%), while the 321 nm band arises mainly from a HOMO-1 → LUMO transition (64%). These features indicate that –COOH addition enhances the intramolecular charge transfer character by promoting electron flow toward the carboxyl group, which acts as an electron acceptor .
The modification also reduces the HOMO–LUMO energy gap (from -5,58/-1,29 eV to -5,88/-1,66 eV), contributing to light absorption at longer wavelengths. A smaller bandgap and stronger intramolecular charge transfer are favorable characteristics for improving dye performance in DSSCs .
Overall, the computational results successfully reproduce experimental spectral behavior and confirm that carboxylation enhances electronic conjugation and transition directionality. γ-Mangostin+COOH therefore possesses greater potential as a natural sensitizer than native γ-mangostin.
3.3. Charge-Transfer Characteristics
Charge-transfer behavior of γ-mangostin and γ-mangostin+COOH was evaluated using key photovoltaic parameters including ΔGinject (electron-injection spontaneity), ΔGregn (driving force dye regeneration), τ (excited-state lifetime), |VRP| (electronic coupling constant), and LHE (light harvesting efficiency). The results are summarized in Table 2.
Calculated charge-transfer parameters
Dye | ΔGinject, eV | ΔGregn, eV | τ, ns | |VRP|, eV | LHE |
γ-mangostin | -3,30 | -0,95 | 5,80 | 0,82 | 0,32 |
γ-mangostin+COOH | -2,85 | -1,21 | 3,01 | 0,96 | 0,53 |
γ-Mangostin exhibits a ΔGinject of -3,30 eV, whereas γ-mangostin+COOH shows a value of -2,85 eV. Both negative values indicate spontaneous electron injection into TiO₂ . Although γ-mangostin has a slightly more negative value, the reduction in magnitude for the –COOH derivative may indicate a more balanced injection process with lower energy loss as heat. Both molecules exhibit a spontaneous driving force for dye regeneration ΔGregn, with values of -0,95 eV for γ-mangostin and -1,21 eV for γ-mangostin+COOH.
Excited-state lifetimes decrease from 5,80 ns (γ-mangostin) to 3,01 ns (γ-mangostin+COOH), while the electronic coupling constant |VRP| increases significantly from 0,82 eV to 0,96 eV. The shorter excited-state lifetime (τ) and increased electronic coupling |VRP| parameter observed for γ-mangostin+COOH suggest a molecular structure that is more favorable for electron-transfer processes relevant to DSSC operation . Although explicit dye–TiO₂ adsorption was not computed, the presence of a carboxyl group is widely recognized as an effective anchoring functionality in practical DSSC systems. Therefore, the observed electronic trends indicate that γ-mangostin+COOH is structurally better suited for surface attachment and interfacial charge transfer, a hypothesis that should be further validated through explicit interfacial modeling or experimental studies.
The Light Harvesting Efficiency (LHE) exhibits a substantial improvement, rising from 0,32 to 0,53. This enhancement is further supported by the broader absorption spectrum of γ-mangostin+COOH, as illustrated in Figure 3. Consequently, the γ-mangostin+COOH derivative generates a larger oscillator strength (f), leading to a higher LHE and a more efficient light-harvesting capability
Collectively, the improved |VRP| and LHE values indicate that γ-mangostin+COOH possesses electronic characteristics favorable for DSSC sensitization. However, explicit interfacial electron-transfer processes at the dye–TiO₂ interface were not directly modeled and should be confirmed through future interfacial simulations or experimental studies.
4. Conclusion
Theoretical oxidation of the prenyl group in γ-mangostin to introduce a carboxyl (–COOH) functionality modifies its electronic structure in a manner favorable for DSSC sensitization. The substitution narrows the HOMO–LUMO gap, enhances light-harvesting efficiency, and improves electronic descriptors associated with charge-transfer processes. Although explicit dye–TiO2 interfacial interactions were not modeled, the molecular electronic trends suggest that γ-mangostin+COOH is structurally better suited for surface anchoring in practical DSSC systems. These findings provide a theoretical foundation for future interfacial simulations and experimental validation.